This article discusses different aspects of thalassemia – a congenital disorder. Here, learn about the types, tests, prevention, and more.
Keywords: Thalassaemia |α globin chain|β-globin chain|delta chain | gamma chain |hemoglobin | Cooley | Mediterranean anemia|multi-organ failure | |
Table of contents
| 1. | Introduction |
| 2. | Classification |
| 3. | Recent advances |
| 4. | Clinical |
| 5. | Links |
Know about’ learn and fly.co.in .’
This article is part of my mission to provide trustworthy recent health information to support the general public, patients, and professionals globally. Here you will find human Physiology, Anatomy, and health-related topics.
Introduction
Thalassemia is a congenital blood disorder due to a defect in hemoglobin synthesis. Due to abnormal hemoglobin, red blood cells are destroyed prematurely, leading to anemia. Thalassemia was first used in 1932. Before that, it was known as Mediterranean anemia. As the disease was first recognized around the Mediterranean sea.
World thalassemia day is May 8.
According to the ICD classification, thalassemia is D-56.
Β major-thalassemia is also known as Cooley anemia.
Thalassemia is an autosomal recessive disease like sickle cell anemia.
The genes are responsible for healthy hemoglobin production. The beta globin chain is encoded by a single gene on the short arm of chromosome11, while the alpha chain encodes two closely linked genes on the short arm of chromosome 16. Each cell contains four copies of the alpha globin gene, and each gene produces one-fourth of the alpha gene.
Normal HbA has gene αα/ αα
silent carrier of α-thalassemia has gene – α /αα
Mild thalassemia has –/αα gene
Major has – – / – α gene
Barts hydrops fetalis, all four genes are absent, so α chains are not produced; only beta chains are formed. Beta chains group in four; this is hemoglobin H (HbH).
Two alpha and two beta chains are present in adult hemoglobin (HbA). Two alpha and delta chains are present in adult hemoglobin (HbA2). Two alpha and two gamma chains are in fetal hemoglobin (HbF).
Normal HbA has 2 alpha and 2 beta chains. Alpha globin protein level equals beta globin protein level.
HbF has 2 alpha and 2 gamma chains, while HbA2 has 2 alpha and 2 delta chains. A single gene on chromosome11 encodes for beta, gamma, delta, and other chains.
Types of thalassemia
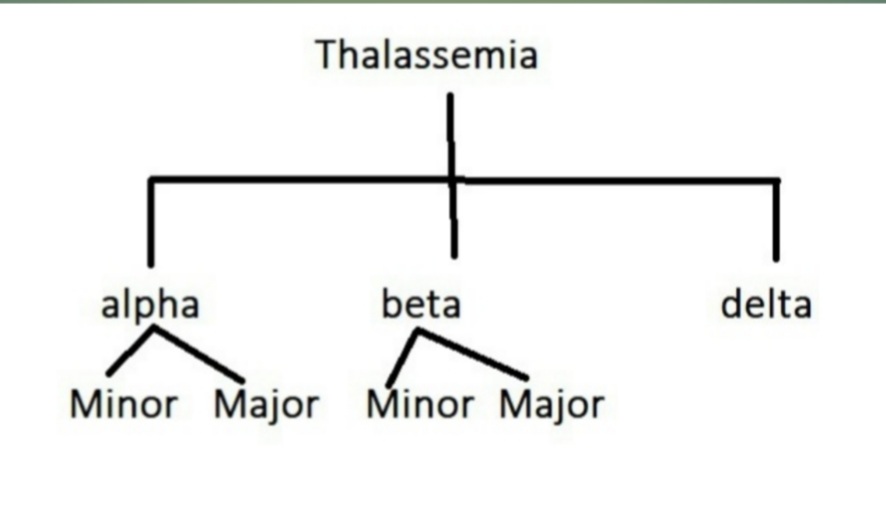
Depending on the presence of a defect in the globin chain of the hemoglobin, thalassemia is classified as
- α -thalassemia -the fault is in the α chain of the hemoglobin. When α-chain is absent, it is known as α – major thalassemia; when reduced, it is α -minor thalassemia.
2. β thalassemia -the defect is in the β -chain of the hemoglobin. When β -the chain is absent, it is known as β-major thalassemia, and when reduced, it is β -minor thalassemia. Intermediate beta thalassemia is also present. The beta globin chain is encoded by a single gene on the short arm of chromosome11. Each cell contains two copies of the beta-globin chain. The beta globin chain is encoded by a single gene on the short arm of chromosome11. β gene also codes for the gamma chain. Therefore,β-thalassemia is not apparent up to eighteen months of age due to fetal hemoglobin. An excess of α chains causes unpaired α-chains.
The beta globin chain is encoded by a single gene on the short arm of chromosome11,
The beta globin chain is encoded by a single gene on the short arm of chromosome11; β-major thalassemia is the most common type of thalassemia.
In normal HbA, b/b genes are present. In minor β- thalassemia b /- (one gene is present, another is absent, while in major -/-, no genes are present on the short arms of chromosome 11.
3. Delta thalassemia is also present as 3% of adult hemoglobin (HbA2) has a delta chain instead of a beta chain. Therefore, gene mutation to produce delta chains is responsible for delta thalassemia.
Thalassemia may coexist with other defects of hemoglobin. For example, it may coexist with sickle cell disease or sickle cell trait.
Thalassemia protects against malaria to some extent.
Incidence: About 280 million people were suffering globally, of which 4.4 lakhs were seriously ill—in 2015. About 7% of the world’s population is suffering from thalassemia. Incidence in India is about one million cases.
Thalassemia is very common in Italy, Greece, South Asia, the middle east, and Africa.
Sex : males: females ratio is 1:1(similar disease rates.).
The life span of an adult red blood cell is 120 days. However, in infants, the life span of red blood cells is less than 100 days.
About 2.4 million new erythrocytes are formed per second in humans.
Diagnosis:
History – Family history, history of blood transfusion, failure to thrive, and lethargy.
Physical examination: In mild cases, history will suggest the diagnosis; otherwise, one may miss the diagnosis.
The red blood cell has some enzymes for glycolysis and hexose monophosphate shunt to get energy.
Signs and symptoms:
In mild thalassemia, there are no symptoms. This may start insidiously. However, symptoms may be severe even to endanger life.
There is mild to severe anemia. Initially, signs and symptoms are due to anemia.
Signs and symptoms of anemia:
If anemia is mild, there is no sign and symptoms, and it goes unnoticed. But when anemia worsens, the symptoms appear and intensify. The symptoms are not specific and include:
1. Lethargy, extreme fatigue.
2. Weakness.
3. Chest pain
4. Headache, dizziness, or lightheadedness
5. Brittle nails
6. Pale skin, palm.
7. Poor appetite
8. Soreness of tongue
Signs are :
Pale skin, in severe cases person, will become paperwhite.
In an early case of anemia, examining the color of the lower conjunctiva and the palm will reveal that they are pale.
Tachycardia: fast heartbeat and rapid pulse rate.
Dyspnoea –shortness of breath.
Cold hands and feet.
Inflammation of gum and tongue.
Brittle nails.
Growth retardation in children.
Abdomen examination: splenomegaly(enlarged spleen) may be present.
Excessive iron accumulates in the body due to the rapid destruction of red blood cells and frequent blood transfusions. In thalassemia, iron deposits in the liver, heart, and other systems, including the endocrine system. This iron deposit damages the functions of these tissues and may be fatal. Therefore, the iron chelating agent is given to remove irons from the tissues.
Excessive red blood cell formation results in abnormal bone structure, especially in the face and skull. As a result, bone marrow expands, and the bone becomes brittle and may fracture easily. Maxillary overgrowth (chipmunk ), malocclusion of teeth, frontal bossing, chronic sinusitis.
X-ray skull shows generalized skeletal osteoporosis, hair on end appearance.
X-ray ribs, vertebrae, metacarpal bones, etc., shows thinning of the cortex. Therefore, bones become brittle.
The spleen enlarges due to the destruction of red blood cells. An enlarged spleen makes anemia worsen.
Cardiac problems may lead to heart failure and abnormal cardiac rhythm.
Increased incidence of infection occurs in thalassemia.
In thalassemia, the growth rate decreases, which is apparent in children. This is due to defective secretion of growth hormone.
In suspected cases, prenatal testing may diagnose the condition.
Diagnosis of thalassemia is confirmed by complete blood count, special hemoglobin investigation-electrophoresis, high-performance liquid chromatography, and genetic test. Serum ferritin, red cell phenotyping, DNA analysis, and LFT are diagnostic tools.
Complete blood count -Microcytic (small red blood cells), hypochromic RBC.Microcytosis without an increase in RDW.
Peripheral blood film examination shows reticulocytosis.
Osmotic fragility decreases. The urine investigation shows increased urobilinogen.
Stool examination shows increased stercobilinogen.
Mentzer index indicates thalassemia. It is calculated from the complete blood count. It is MCV( mean corpuscular volume) per red cell count. If it is less than 13, it suggests a thalassemia trait. And if the value is more than 13, it is due to iron deficiency anemia.
Rapid hemolysis and repeated blood transfusion cause iron accumulation in different organs and affect their functions. Iron is deposited in the liver, heart, endocrine glands, etc. Features of hypothyroidism, hypoparathyroidism, hypopituitarism and insulin deficiency occur.
Complications of repeated blood transfusions, for example, infections of Hepatitis B, C, D, parvovirus B19, etc.
Prevention is better than cure; therefore, it is recommended that all couples must be tested for thalassemia -genetic testing and counseling.
Treatment: Medical treatment is not essential in mild thalassemia. In severe disease:
Repeated blood transfusion, iron chelation, and folic acid are the mainstay of treatment.
Indication of blood transfusion -Blood transfusion is given when Hemoglobin finding is below 7 gm /dl in two tests performed two weeks apart. Blood transfusion is given at an interval of four weeks to maintain hemoglobin at 9-10 gm/dl, and post transfusional hemoglobin level is 13.5 to 15.5 gm/dl.
Growth hormone replacement therapy is helpful in the expected growth of children.
A bone marrow transplant may be done.
For iron chelation -deferoxamine, deferiprone ., etc. are used.
FDA-approved drug is Hydroxyurea. However, thalidomide is also effective and is used in combination with Hydroxyurea.
Life expectancy is 50 -60 years in managed cases.
Cause of death -Congestive heart failure, arrhythmia, and /or multi-organ failure.
Summary
Thalassemia is not a curable disease, but treatment is available. Life expectancy is normal with proper treatment.
This article will be helpful for you.
Like to meet again? Sign up to get an email when I publish.
Question? Email me bkp337390@gmail.com
Follow me on Instagram. (https://www.instagram.com/
Andon’t’t forgets to follow me on learn and fly .co.in.
I am more enthusiastic, and you can get new insights from my articles later.
Thank you.
Disclaimer: All possible measures have been taken to ensure the accuracy and reliability of the information; however, ‘learn and fly.co.in’ does not take any liability for the same using any information provided by the website is solely at the viewers.’The information is provided as an educational service and public awareness. It is not medical advice.
In case of any medical health issue, we advise you to seek the advice of a qualified doctor and follow his instructions.
Learn More :
1. About us
2. Contact us
3. Disclaimer
4. Privacy Policy
Please submit any comments about this article. The team will work hard to evaluate the statement and make appropriate corrections.
Help to improve the content.
Question: Hi! Got any questions? I can help you.
Have a comment about this article. Please let me know.
Your name:
Email:

This Photo is by Sandhya Prasad, Licensed under CCBY.
Internal link: http://blog.totalphysiology.com/2020/12/erythrocytes-rbc.html
https://www.totalphysiology 20.blogspot.com/2021/01/anemia.html
External link: https://en.m.wikipedia.org/wiki/Thalassemia

very informative
LikeLike
excellent article,Sir
LikeLike